Java Snippets
Most of the example on this page are related to the CDK
package, an opensource framework for cheminformatics. The
examples are mainly for my utility (so that I can see what
I did 6 months ago!) but they do show various aspects of CDK
functionality as well as Java programming and so might
be useful to other people as well. Much of the code is
based of code from the JUnit tests so thanks goes to the
CDK
developers for a well designed and documented framework.
NOTE: Nightly builds are now available
Miscellaneous Utilities | Pharmacophore Searching | Building & Testing | Reading conformer data | 3D similarity | Fragmenting a molecule | SMILES to SDF | Molecular Descriptor GUI | Surface areas in the CDK | Geometric transformations | Reading molecules from disk | Writing molecules to disk | Calculating molecular descriptors | CDK as a web service | Maximum Common Substructures (MCSS) | "Uncommon" Substructures | Shortest path length matrix | Tabular display of 2D structures | Batch generation of 2D diagrams
You can get guhautils.jar and Javadocs. Right now there are relatively few methods, mainly focused on loading files and depictions. The latter is slightly problematic as the current CDK 1.2.x release candidate removed depictions. As a result, I provide a modified version of the latest 1.2.x branch that contains the work from the latest jchempaint-primary branch (as of 23rd February). You can get the jar file here. Unfortunately, due to the active development in JChemPaint, the depiction code is slightly broken since the molecule edges appear to be cutoff.
An exmple of depicting a SMILES string is
import net.guha.util.cdk.Misc; import net.guha.util.cdk.Renderer2DPanel; import org.openscience.cdk.DefaultChemObjectBuilder; import org.openscience.cdk.interfaces.IAtomContainer; import org.openscience.cdk.smiles.SmilesParser; import javax.swing.*; public class SimpleDepiction { public static void main(String[] args) throws Exception { SmilesParser smilesParser = new SmilesParser(DefaultChemObjectBuilder.getInstance()); String smiles = "c1cc(CC=CC#N)ccn1"; IAtomContainer molecule = smilesParser.parseSmiles(smiles); molecule = Misc.get2DCoords(molecule); Renderer2DPanel rendererPanel = new Renderer2DPanel(molecule, 200, 200); rendererPanel.setName("rendererPanel"); JFrame frame = new JFrame("2D Structure Viewer"); frame.setDefaultCloseOperation(JFrame.DISPOSE_ON_CLOSE); frame.getContentPane().add(rendererPanel); frame.setSize(200, 200); frame.setVisible(true); } }If you'd like to highlight a substructure, it's just a little extra work. For example, we can use a SMARTS pattern to identify and retrieve the substructure of a molecule and then show the depiction with it highlighted. The key thing to remember when highlighting substructures, is that the atoms in the needle molecule should be the same as the atoms in the target molecule. That is, do not create new atoms from the original molecule, but simply add them to the needle molecule.
import net.guha.util.cdk.Misc; import net.guha.util.cdk.Renderer2DPanel; import org.openscience.cdk.DefaultChemObjectBuilder; import org.openscience.cdk.interfaces.IAtomContainer; import org.openscience.cdk.smiles.SmilesParser; import javax.swing.*; public class SimpleDepiction { public static void main(String[] args) throws Exception { SmilesParser smilesParser = new SmilesParser(DefaultChemObjectBuilder.getInstance()); String smiles = "c1cc(CC=CC#N)ccn1"; IAtomContainer molecule = smilesParser.parseSmiles(smiles); molecule = Misc.get2DCoords(molecule); Renderer2DPanel rendererPanel = new Renderer2DPanel(molecule, 200, 200); rendererPanel.setName("rendererPanel"); JFrame frame = new JFrame("2D Structure Viewer"); frame.setDefaultCloseOperation(JFrame.DISPOSE_ON_CLOSE); frame.getContentPane().add(rendererPanel); frame.setSize(200, 200); frame.setVisible(true); } }
Update 12/7/11: Output now includes similarity values along with the title for the matching molecule
Update 12/3/11: Updated to process multiple input files specified on the command line
Update 11/22/11: Updated to use the latest CDK master. Sources are now available in Github
Recently Bellester et al described a descriptor based method (as opposed to superposition methods) for 3D similarity searches. The work is reviewed here. The nice thing about the method is that it's quite fast and space efficient.
THe method has been implemented in the CDK as the DistanceMoment class. The class allows you to calculate the 3D similarity between two molecules as well as generate the distance moments for a given molecule. Note that the class does not perform hydrogen removal, so this must be done by the caller.
Informal testing indicates that it takes 0.3 seconds per 1000 structures (this includes reading and writing to disk), so theres definite room for improvement and some proper benchmarking. You can try out momsim.jar which allows you to generate the distance moment vectors as well as search a target SD file, given a query and similarity cutoff. It includes all the relevant CDK classes so it should be standalone. Simple usage is
java -jar momsim.jar -v -g somemols.sdfThis will generate the 12-D vectors for each molecule in somemol.sdf, writing the output to mom.txt, though this can be changed on the command line. If 3D coordinates are missing a 12-element vector of NA's is generated. The program will not evaluate the distance vectors for molecules with 1 heavy atom. Do java -jar momsim.jar -h to get a description of the other parameters available
- To recompile all the tests do
ant test-dist-all
- To test a specific class
ant -Dtestclass=qsar.DescriptorEngineTest junit-test
- To test a module
ant -Dmodule=MODULENAME test-module
- To aovid running slow tests as -DrunSlowTests=false
- To compile a source module
ant -Dmodule=atomtype -Dsource=src/main compile-module
- To compile a test module
ant -Dmodule=test-atomtype -Dsource=src/test compile-module
The CDK now supports reading and storing conformers efficiently. The IteratingMDLConformerReader allows one to iterate over the conformers of a molecule stored in SD format. It assumes that all conformers for a given molecule have the same title (a graph isomorphism test is planned for the future). Thus rather than getting a molecule object at each iteration we now get back a collection of conformers at each iteration.
The conformers for a molecule are stored in an object of class ConformerContainer. This is a memory efficient data structure and is modeled on a List. Thus one can iterate over the conformers or retrieve a specific conformer. Example usage of these classes is
String filename = "/Users/rguha/conf2.sdf"; IteratingMDLConformerReader reader = new IteratingMDLConformerReader( new FileReader(new File(filename)), DefaultChemObjectBuilder.getInstance()); while (reader.hasNext()) { ConformerContainer cc = (ConformerContainer) reader.next(); int i int nconf = 0; for (IAtomContainer conf : cc) { // do something with this conformer nconf++; } }
Usage is simply
java -cp $CLASSPATH:./ smi2sdf "CCC=C=CO"By default, the resultant SD file is named output.sdf though this can be changed on the command line. Note that you will need the Apache Commons CLI package in the classpath. (This is included in the CDK distribution)
- The probe radius
- The level of tesselation
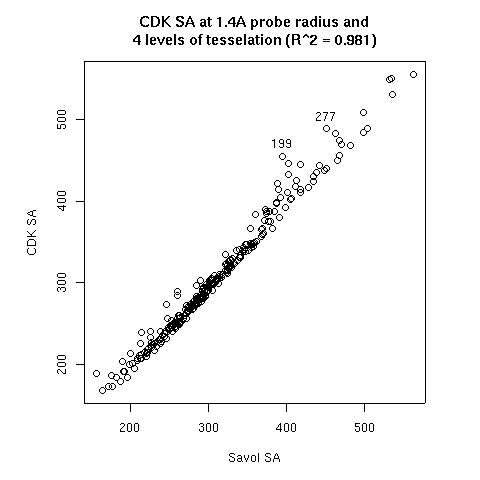
|
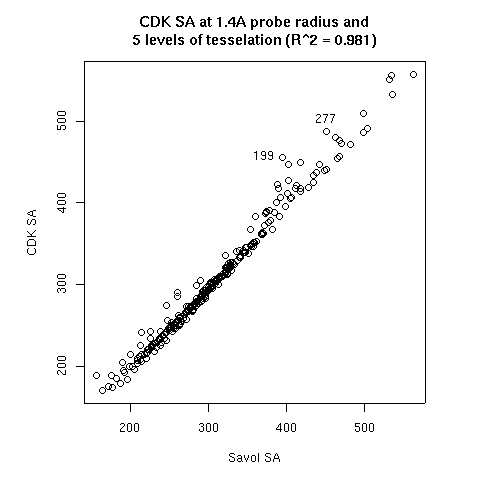
|
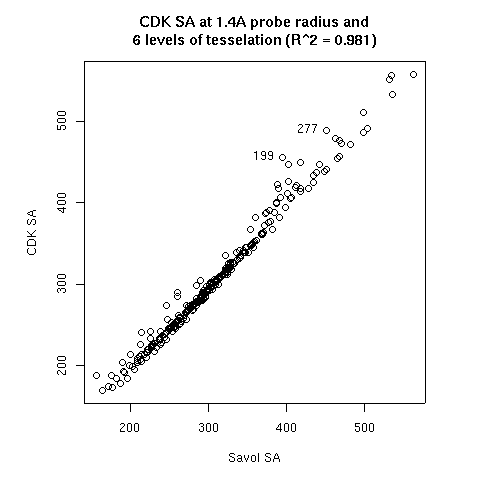
|
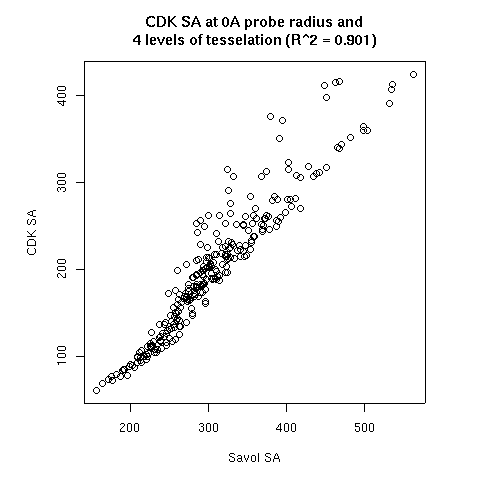
|
- rotation: rotate.java rotates a structure about the X, Y or Z axes by a specified angle. The center of rotation is the origin. Currently, the program will only rotate a single structure.
- translate: translate.java translates a structure by specified amounts along the X, Y and Z axes. In addition if specified, the center of gravity of the structure can be translated to the origin.
CDKPsearch-1.3.0.jar is a standalone application (source code) that allows you to perform a pharmacophore search on a collection of molecules stored in SD format. The program accepts an SD file with single (i.e., one conformer) structures or multi-conformer structures. In the latter case, conformers are detected based on titles. Thus all conformers for a given molecule should be located in sequence and should have the same title. The program will write out the structures that match the query to the file hits.sdf and also provide a summary report in report.txt. You can run the program as
java -jar CDKPsearch-1.3.0.jar --sdfile targets.sdf --query query.xml -cIf targets.sdf is a not conformer collection you can remove the -c parameter, though it will probably not make a difference (except a slight drop in speed). Run the program with no arguments to get a help page.
The format for the pharmacophore query file is XML with a RelaxNG schema available here. This can be used to validate any pharmacophore definition file using tools such as jing. An example of a query file containng multiple pharmacophore queries is here. Note that the CDKPsearch application assumes that a query file will have a single query and if multiple queries are present will complain. This will be updated in the future. The schema supports both distance and angle constraint, both are currently supported by the CDK. Dihedral constraints are on the way.I would welcome any suggestions or comments on the schema with the aim of making it a common way to exchange pharmacophore queries between multiple systems.
Update (14/04/2015) The project is mavenized and updated to CDK 1.5.0
Update (23/05/2012) The --validate option will validate an query file to ensure that it is correctly specified according to the RelaxNG schema. Currently does not validate the SMARTS patterns in the query
Update (22/05/2012) Updated to latest stable CDK version. Updated so that conformer mode also outputs dummy atom annotated hits
Update (27/01/2012) Updated to CDK 1.4.7
Update (15/07/2008) Now a query file can contain multiple query definitions. You can choose which one to use by specifying the name of the query with the --qname argument. If --qname is not specified, the first query definition in the qery file is used
Update (09/07/2008) Minor update to take into a SMARTS bug fix (related to ring counts in recursive SMARTS) in CDK trunk
Update (24/06/2008) Updated to the latest CDK trunk. Also now has support
for pharmacophore group SMARTS which use a logical OR operator to join one or more
multi-atom SMARTS (as opposed to traditional OR which only allows single atom matches).
Use the | symbol to separate multiple SMARTS in a group definition
Update (19/06/2008) Updated to be a little more robust if input files
are missing. Default hit file name is now a combination of the query and input SD
file names. Also updated to support angle constraints in the detailed output
Update (11/06/2008) Updated to the latest CDK trunk so that it now supports
angle constraints
Update (21/05/2008): Updated to check whether a molecule has 3D coordinates. If so, skip the molecule
Update (20/05/2008): Added the -a parameter, which will cause the molecules in
the output file containing the hits, to be annotated with pharmacophore groups. This
is achieved by placing a Xe atom at the coordinates for each pharmacophore group
of the query (only considering those groups in the target that satisfied the query). This
means that multi-atom groups will have a Xe atom at their centroid (eg., phenyl rings)
whereas for single-atom groups, the original atom will be hidden by the Xe atom. This is
a very crude way to visualize pharmacphores (say in Jmol), especially since all the groups are of the same
size and color (though the bond colors don't get changed, so that gives some indication of
what a hidden atom actually was).
UPDATE (13/05/2008): Updated to avoid notification messages on core
CDK objects. 2.3x speedup! Timing is also reported when verbose output is
selected. Also updated to provide more detail if desired. With the -d option,
the report file will include details of the distance constraints that matched in
a target. Right now, since we just support distance contraints, each match is a
3-tuple of the form (G1, G2, dist), where G1 and G2 are names of pharmacophore groups
UPDATE (07/05/2008): Synced with the latest CDK
UPDATE (04/04/2008): Synced with the latest CDK, so better aromaticity detection
UPDATE (31/10/2007): Updated to the latest CDK, so should have better SMARTS matching
String filename = "molecules.sdf"; InputStream ins = this.getClass().getClassLoader().getResourceAsStream(filename); MDLReader reader = new MDLReader(ins); // alternatively, you can specify a file directly // MDLReader sdfreader = new MDLV2000Reader(new FileReader(new File(filename))); ChemFile chemFile = (ChemFile)reader.read((ChemObject)new ChemFile()); List containersList = ChemFileManipulator.getAllAtomContainers(chemFile);You could replace MDLReader with a variety of other readers for diferent formats (see here). Note that if you're trying to read multiple files at one go from an SD file, you should MDLV2000Reader rather than MDLReader. At the end containersList would contain all the molecules stored in the file as IAtomContainer objects.
Howver a more general code snippet below allows you to simply specify the filename and automatically detect the format and load the molecules
public static IAtomContainer[] loadMolecules(String[] filenames) throws CDKException { Vector v = new Vector(); DefaultChemObjectBuilder builder = DefaultChemObjectBuilder.getInstance(); try { int i; int j; for (i = 0; i < filenames.length; i++) { File input = new File(filenames[i]); ReaderFactory readerFactory = new ReaderFactory(); IChemObjectReader reader = readerFactory.createReader(new FileReader(input)); IChemFile content = (IChemFile) reader.read(builder.newChemFile()); if (content == null) continue; List c = ChemFileManipulator.getAllAtomContainers(content); // we should do this loop in case we have files // that contain multiple molecules for (j = 0; j < c.size(); j++) v.add((IAtomContainer) c.get(j)); } } catch (Exception e) { e.printStackTrace(); throw new CDKException(e.toString()); } // convert the vector to a simple array IAtomContainer[] retValues = new IAtomContainer[v.size()]; for (int i = 0; i < v.size(); i++) { retValues[i] = v.get(i); } return retValues; }It should be noted that loading molecules from disk follows the principle of "least surprise", in that the methods anly load the molecule and do not do any more processing. Thus aromaticity detection is not performed automatically and you have to use the HueckelAromaticityDetector class.
Also note that the CDK has been refactored to use NULL default values for many atom specific variables. This allows us to differentiate between something that was set to a default of 0 and something that is actually set to 0 by some method. In general, it's a good idea to check for a NULL (represented by CDKConstants.UNSET)
To write a Molecule object to a String in the MDL MOL format you can do
import org.openscience.cdk.io.MDLWriter; import java.io.StringWriter; StringWriter w = new StringWriter(); try { MDLWriter mw = new MDLWriter(w); mw.write(ac); mw.close() } catch (Exception e) { System.out.println(e.toString()); } System.out.println(w.toString());Alternatively to write it to a file you can do
import org.openscience.cdk.io.MDLWriter; import java.io.FileWriter; import java.io.File; FileWriter w = new FileWriter(new File("molecule.mol")); try { MDLWriter mw = new MDLWriter(w); mw.write(ac); mw.close(); } catch (Exception e) { System.out.println(e.toString()); }
IAtomContainer ac; IDescriptor descriptor = new WienerNumbersDescriptor(); DoubleArrayResult retval = (DoubleArrayResult)descriptor.calculate(ac); double wpath = retval.get(0); // Wiener path number double wpol = retval.get(1); // Wiener polarity numberIt should be noted that the return values for descriptors can be single integers or doubles or arrays of integers or doubles or boolean values. The return values are objects that implement the DescriptorResult interface. A general method of obtaining the return values (without coercion) is
DescriptorResult retval = descriptor.calculate(ac); if (retval instanceof DoubleArrayResult) { // process } else if (retval instanceof DoubleResult) { // process } ...In some cases, descriptor routines will take parameters. An example is the BCUT descriptor. In this case the user can specify how many eigenvalues should be returned. This can be done by the following code
IAtomContainer ac; IDescriptor descriptor = new BCUTDescriptor(); Object[] params = { new Integer(3), new Integer(3) }; descriptor.setParameters(params); DoubleArrayResult retval = (DoubleArrayResult)descriptor.calculate(ac);The above code returns the 2 highest and 2 lowest eigenvalues of the weighted Burden matrix.
In addition to the calculation of individual descriptors it is also possible to evaluate all descriptors or subsets of descriptors implemented in the CDK. This is performed by the DescriptorEngine. To calculate all available descriptors we can use the code below
Molecule molecule; // initialize the Molecule object DescriptorEngine engine = new DescriptorEngine(); engine.process(molecule);In case we want to calculate specific classes of descriptors (say topological and geometric) we can do
String[] types = {'topological','geometric'}; DescriptorEngine engine = new DescriptorEngine(types); engine.process(molecule);In both cases, the result of each descriptor is stored in the Molecule object as a DescriptorValue object keyed on the DescriptorSpecification object for that descriptor (which contains, among other things, a link to a metadata dictionary entry for that descriptor).
int[][] admat = AdjacencyMatrix.getMatrix(atomContainer); int[][] m = PathTools.computeFloydAPSP(admat);m is the pairwise distance matrix. A program that computes this matrix and displays it can be obtained here
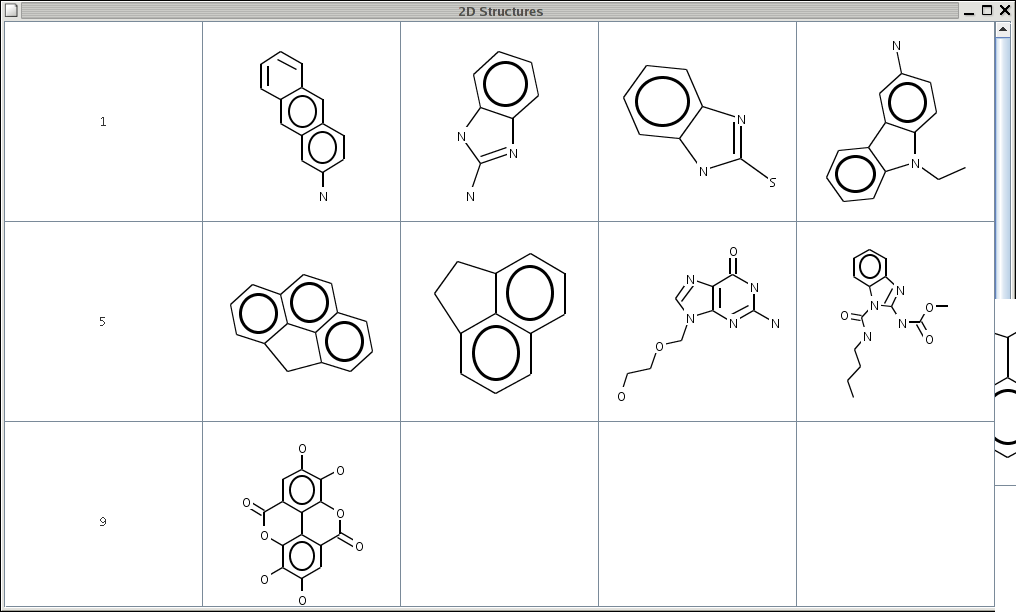{kind=link}
The code allows the user to specify 4 properties
- ncol - the number of columns to display
- cellx - the width of each cell in the table
- celly - the height of each cell in the table
- withH - a boolean to indicate whether hydrogens should be shown or not
java -Dncol=5 st2d dan*.hinand
java -Dncol=4 -Dcellx=300 -Dcelly=400 -DwithH=true s2td molecule*.cmlRight now the molecules are drawn within a fixed cell size. That is, if the table is resized, the molecules do not get redrawn for the new cell size. Currently I'm not sure on how to handle this. Another nice thing would be to be able to set arbitrary tooltips for each cell.
The MCSS is C1CC1. In the case of the first molecule, there are two disconnected substructures that are not part of the MCSS, whereas in the second case there will be two. Thus given two molecules, whose MCSS has been determined, we would like to get a list of substructures from each molecule that are not part of the MCSS. The following code snippet shows the approach:
List matches = UniversalIsomorphismTester.getSubgraphAtomsMaps(mol, mcss); List mapList = (List) matches.get(0); for (Object o : mapList) { RMap rmap = (RMap) o; atomSerialsToDelete.add(rmap.getId1()); } ArrayList atomsToDelete = new ArrayList(); for (Integer serial : atomSerialsToDelete) { atomsToDelete.add(mol.getAtom(serial)); }Here we do a subgraph matching to identify which atoms in our molecule correspond to those identified in the MCSS. Note that the subgraph matching returns serial numbers. To safely handle deletions, it is best to get the actual atoms that we will delete. Given a list of atoms, it is easy to delete them and their associated bonds:
for (IAtom atom : atomsToDelete) {
mol.removeAtomAndConnectedElectronContainers(atom);
}
Finally, since in general the resultant molecule will now have disconnected components we
would like to get back set of molecules, each one representing one of the components
return ConnectivityChecker.partitionIntoMolecules(mol);The complete code that can be run from the command line is in NoMCSS.java.
List mcsslist = UniversalIsomorphismTester.getOverlaps( atomcontainer1, atomcontainer2 ); int maxmcss = -9999999; IAtomContainer maxac = null; for (i = 0; i < mcsslist.size(); i++){ IAtomContainer a = (IAtomContainer)mcsslist.get(i); if (a.getAtomCount() > maxmcss) { maxmcss = a.getAtomCount(); maxac = a; } }Once a MCSS is found, we would like to display the molecules with the MCSS portion highlighted. To map the the MCSS to each molecule we must determine which bonds of the MCSS map to which bonds in the original molecules. This is effectively a substructure search in which the MCSS is the query fragment .This is performed by using the UniversalIsomorphismTester.getSubgraphMaps() function (see highlighting substructures for a more detailed description of this process). This function can return multiple mappings (for instance if the MCSS is a benzene ring then anthracene would have three mappings for this MCSS). The code below only takes the first mapping
// a is a molecule from the set of molecules for which a MCSS was obtained // and q is the MCSS which is obtained as shown above public static IAtomContainer getneedle(IAtomContainer a, IAtomContainer q) { IAtomContainer needle = DefaultChemObjectBuilder.getInstance().newAtomContainer(); Vector idlist = new Vector(); List l = UniversalIsomorphismTester.getSubgraphMaps(a, q); List maplist = (List)l.get(0); for (Iterator i = maplist.iterator(); i.hasNext(); ) { RMap rmap = (RMap)i.next(); idlist.add( new Integer( rmap.getId1() ) ); } HashSet hs = new HashSet(idlist); for (Iterator i = hs.iterator(); i.hasNext();) { needle.addBond( a.getBondAt( ((Integer)i.next()).intValue() ) ); } return needle; }The return value of this function is an IAtomContainer containing the bonds from a that correspond to the bonds in q (the MCSS). The return value is then used to specify which bonds in the molecule a are to be highlighted with the Renderer2D class. A small program that combines all these features is simplemcss.java which determines and displays the MCSS for two molecules highlighted on each molecule. You can see screenshots here and here. To use it compile as usual and then do
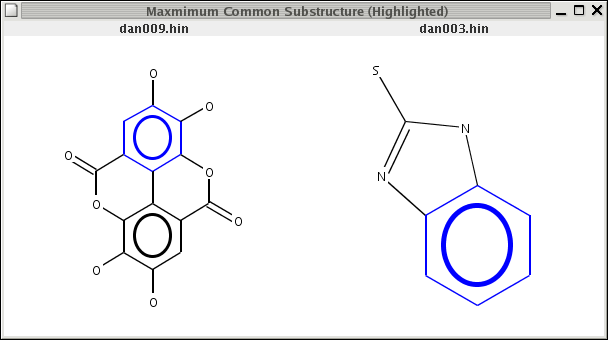{kind=link}
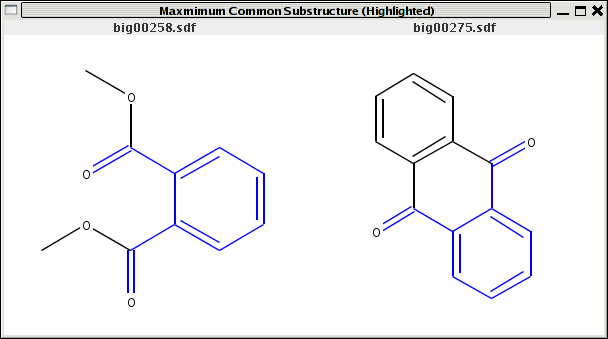{kind=link}
java simplemcss molecule1.hin molecule2.hin(replace the molecule files with whatever you have).
The Fragmenter2 class performs this operation and given a molecule it will generate an ArrayList of IAtomContainter's, each one representing an individual fragment. Fragments are generated by splitting the molecule at
- non-terminal bonds
- non-ring bonds
- at a bond
- or at an atom
In addition to generating the fragments the code will bring up a tabular display of the original molecule (red border) and images of the fragments generated (screenshot).
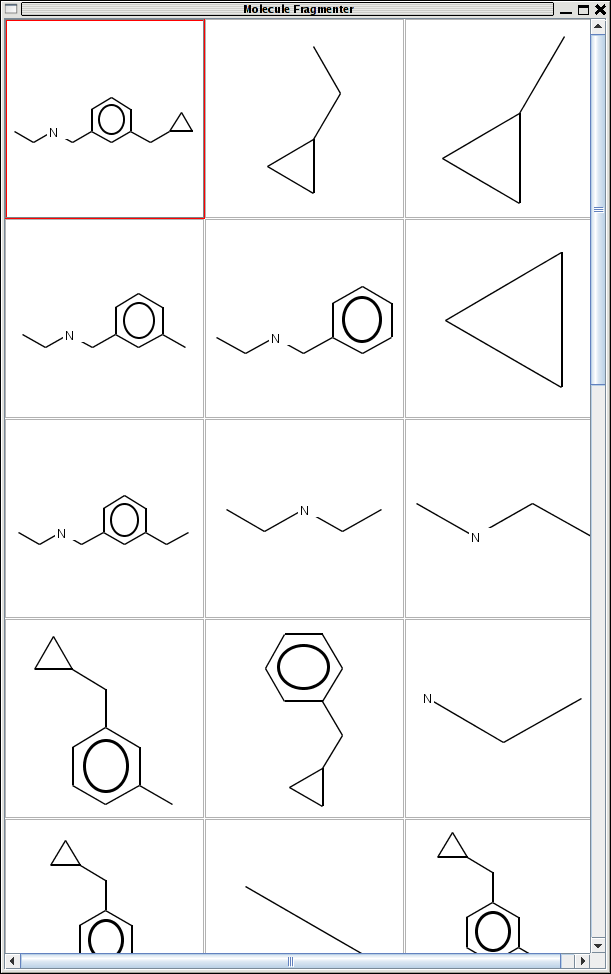{kind=link}
To run it, assuming you have the CDK jar files in your CLASSPATH
javac -cp $CLASSPATH:./ Fragmenter2.java java -cp $CLASSPATH:./ Fragmenter2 SMILESIf no SMILES string is specified it will proceed to fragment c1c(CC2CC2)cc(CNCC)cc1, otherwise it will fragment the specified molecule.
Update: The code has been modified to recursively fragment the initial set of fragments. I think this leads to all possible fragments. The downside is that many of the fragments are topologically identical, but since they involve different atoms of the parent molecule, they get included in the final fragment list. To get a set of topologically unique fragments, we'd have to perform am isomorphism test - which is left to the reader!
Also note that since we recursively fragment the intial set of fragments, if you have long chains as part of your molecule, the program may take a long time to complete.
java -cp $CLASSPATH:./ draw2d molecules.sdfBy default it will generate a series of images named img001.png, img002.png and so on in the current directory. The output directory can be specified. In addition the width and height of the final images can be specified. A scale factor can also be specified that indicates how large the image of the structure will be in comparison to the specified image area.
Requirements: The above usage implies that you have in your CLASSPATH
- iText library
- Relevant CDK jar files
- Libraries that the CDK depends upon
If you are not a developer you can use draw2d.jar which bundles all the required libraries (both the CDK and the libraries it depends upon). Thus you can simply do
java -jar draw2d.jarwhich will show the help message.
Example of tabular PDF's
- Single column of molecules PDF
- 3 molecule columns PDF
- 2 molecule columns with SDF property fields (in this case calculated descriptor values) PDF
usage: draw2d [OPTIONS] sdf_file
Draws 2D images from coordinate files. Default output is PNG and input
should be an SDF file.
-c,--scale Scale factor (default is 0.9)
-f,--format Output format (PNG, JPEG, PDF)
-h,--help Give this help page
-l,--color Color atoms
-n,--ncol How many molecule columns should tabular output have.
The number must be 1 or more
-o,--outputDir Where to dump images (default is current directory)
-p,--props If present output properties in the PDF table. Default
is no
-s,--showH Show explicit hydrogens (default is no)
-t,--table Output structures as a tabular PDF. This option
overrides the format option and the output
file is called output.pdf
-v,--verbose Verbose output
-x,--x Width of the images (default is 300)
-y,--y Height of the images (default is 300)
| Updated (06/05/08) | Updated to the latest CDK, removed dependency on JAI, removed SVG support |
| Updated (08/14/07) | Updated to sync with latest CDK |
| Updated (08/07/06) | Updated to sync with latest CDK. Also provided an option for coloring atoms |
| Updated (05/18/06) | Updated to show H's for heteroatoms |
| Updated (05/10/06) | Updated to be in sync with the latest CDK SVN |
| Updated (04/28/06) | Updated to be in sync with the latest CDK CVS and added explici aromaticity detection |
| Updated (03/20/06) | Updated to be in sync with the latest CDK CVS |
| Updated (02/09/06) | Updated to be in sync with the latest CDK CVS |
| Updated (01/24/06) | Updated to improve the formatting of numbers when the properties are included in tabular PDF output |
| Updated (01/22/06) | The tabular format can now have a user-specifiable number of columns, using the -n argument. Also applied a patch from Egon Willighagen to also output the SDF property values if specified |
| Updated (01/20/06) | Applied patch from Egon Willighagen to produce a PDF table of structures. Also added license text |
| Updated (01/19/06) | Can now produce PDF output. Requires the iText library to be in the CLASSPATH |
| Updated (01/19/06) | Applied patch from Noel O'Boyle to make it work with the latest CDK |